Publications
List of peer-reviewed publications and pre-prints in reverse chronological order.
Poličar, P. G., Stražar, M., & Zupan, B. (2024). openTSNE: A Modular Python Library for t-SNE Dimensionality Reduction and Embedding. Journal of Statistical Software, 109(3), 1–30. | Link | Toggle abstract
Mucosal and barrier tissues, such as the gut, lung or skin, are composed of a complex network of cells and microbes forming a tight niche that prevents pathogen colonization and supports host–microbiome symbiosis. Characterizing these networks at high molecular and cellular resolution is crucial for understanding homeostasis and disease. Here we present spatial host–microbiome sequencing (SHM-seq), an all-sequencing-based approach that captures tissue histology, polyadenylated RNAs and bacterial 16S sequences directly from a tissue by modifying spatially barcoded glass surfaces to enable simultaneous capture of host transcripts and hypervariable regions of the 16S bacterial ribosomal RNA. We applied our approach to the mouse gut as a model system, used a deep learning approach for data mapping and detected spatial niches defined by cellular composition and microbial geography. We show that subpopulations of gut cells express specific gene programs in different microenvironments characteristic of regional commensal bacteria and impact host–bacteria interactions. SHM-seq should enhance the study of native host–microbe interactions in health and disease.
Goldman, S., Wohlwend, J., Stražar, M., Haroush, G., Xavier, R. J., & Coley, C. W. (2023). Annotating metabolite mass spectra with domain-inspired chemical formula transformers. Nature Machine Intelligence, 5(9), 965-979. | Link | Toggle abstract
Metabolomics studies have identified small molecules that mediate cell signaling, competition and disease pathology, in part due to large-scale community efforts to measure tandem mass spectra for thousands of metabolite standards. Nevertheless, the majority of spectra observed in clinical samples cannot be unambiguously matched to known structures. Deep learning approaches to small-molecule structure elucidation have surprisingly failed to rival classical statistical methods, which we hypothesize is due to the lack of in-domain knowledge incorporated into current neural network architectures. Here we introduce a neural network-driven workflow for untargeted metabolomics, Metabolite Inference with Spectrum Transformers (MIST), to annotate tandem mass spectra peaks with chemical structures. Unlike existing approaches, MIST incorporates domain insights into its architecture by encoding peaks with their chemical formula representations, implicitly featurizing pairwise neutral losses and training the network to additionally predict substructure fragments. MIST performs favorably compared with both standard neural architectures and the state-of-the-art kernel method on the task of fingerprint prediction for over 70% of metabolite standards and retrieves 66% of metabolites with equal or improved accuracy, with 29% strictly better. We further demonstrate the utility of MIST by suggesting potential dipeptide and alkaloid structures for differentially abundant spectra found in an inflammatory bowel disease patient cohort.
CD4+ T cell responses are exquisitely antigen specific and directed toward peptide epitopes displayed by human leukocyte antigen class II (HLA-II) on antigen-presenting cells. Underrepresentation of diverse alleles in ligand databases and an incomplete understanding of factors affecting antigen presentation in vivo have limited progress in defining principles of peptide immunogenicity. Here, we employed monoallelic immunopeptidomics to identify 358,024 HLA-II binders, with a particular focus on HLA-DQ and HLA-DP. We uncovered peptide-binding patterns across a spectrum of binding affinities and enrichment of structural antigen features. These aspects underpinned the development of context-aware predictor of T cell antigens (CAPTAn), a deep learning model that predicts peptide antigens based on their affinity to HLA-II and full sequence of their source proteins. CAPTAn was instrumental in discovering prevalent T cell epitopes from bacteria in the human microbiome and a pan-variant epitope from SARS-CoV-2. Together CAPTAn and associated datasets present a resource for antigen discovery and the unraveling genetic associations of HLA alleles with immunopathologies.
Vatanen, T., Jabbar, K.S., Ruohtula, T., Honkanen, J., Avila-Pacheco, J., Siljander, H., Stražar, M., Oikarinen, S., Hyöty, H., Ilonen, J., Mitchell, C.M. ... & Xavier, R. J. (2022). Mobile genetic elements from the maternal microbiome shape infant gut microbial assembly and metabolism. Cell, 185(26), pp.4921-4936. | Link | Toggle abstract
The perinatal period represents a critical window for cognitive and immune system development, promoted by maternal and infant gut microbiomes and their metabolites. Here, we tracked the co-development of microbiomes and metabolomes from late pregnancy to 1 year of age using longitudinal multi-omics data from a cohort of 70 mother-infant dyads. We discovered large-scale mother-to-infant interspecies transfer of mobile genetic elements, frequently involving genes associated with diet-related adaptations. Infant gut metabolomes were less diverse than maternal but featured hundreds of unique metabolites and microbe-metabolite associations not detected in mothers. Metabolomes and serum cytokine signatures of infants who received regular—but not extensively hydrolyzed—formula were distinct from those of exclusively breastfed infants. Taken together, our integrative analysis expands the concept of vertical transmission of the gut microbiome and provides original insights into the development of maternal and infant microbiomes and metabolomes during late pregnancy and early life.
Vatanen, T., Ang, Q.Y., Siegwald, L., Sarker, S.A., Le Roy, C.I., Duboux, S., Delannoy-Bruno, O., Ngom-Bru, C., Boulangé, C.L., Stražar, M., Avila-Pacheco, J., ... & Xavier, R. J. (2022). A distinct clade of Bifidobacterium longum in the gut of Bangladeshi children thrives during weaning. Cell, 185(23), pp.4280-4297. | Link | Toggle abstract
The gut microbiome has an important role in infant health and development. We characterized the fecal microbiome and metabolome of 222 young children in Dhaka, Bangladesh during the first two years of life. A distinct Bifidobacterium longum clade expanded with introduction of solid foods and harbored enzymes for utilizing both breast milk and solid food substrates. The clade was highly prevalent in Bangladesh, present globally (at lower prevalence), and correlated with many other gut taxa and metabolites, indicating an important role in gut ecology. We also found that the B. longum clades and associated metabolites were implicated in childhood diarrhea and early growth, including positive associations between growth measures and B. longum subsp. infantis, indolelactate and N-acetylglutamate. Our data demonstrate geographic, cultural, seasonal, and ecological heterogeneity that should be accounted for when identifying microbiome factors implicated in and potentially benefiting infant development.
Stražar, M.*, Mourits, V. P.*, Koeken, V. A., de Bree, L. C. J., Moorlag, S. J., Joosten, L. A., ... & Xavier, R. J. (2021). The influence of the gut microbiome on BCG-induced trained immunity. Genome Biology, 22(1), 1-22. | Link | Toggle abstract
The bacillus Calmette-Guérin (BCG) vaccine protects against tuberculosis and heterologous infections but elicits high inter-individual variation in specific and nonspecific, or trained, immune responses. While the gut microbiome is increasingly recognized as an important modulator of vaccine responses and immunity in general, its potential role in BCG-induced protection is largely unknown.
Stool and blood were collected from 321 healthy adults before BCG vaccination, followed by blood sampling after 2 weeks and 3 months. Metagenomics based on de novo genome assembly reveals 43 immunomodulatory taxa. The nonspecific, trained immune response is detected by altered production of cytokines IL-6, IL-1β, and TNF-α upon ex vivo blood restimulation with Staphylococcus aureus and negatively correlates with abundance of Roseburia. The specific response, measured by IFN-γ production upon Mycobacterium tuberculosis stimulation, is associated positively with Ruminococcus and Eggerthella lenta. The identified immunomodulatory taxa also have the strongest effects on circulating metabolites, with Roseburia affecting phenylalanine metabolism. This is corroborated by abundances of relevant enzymes, suggesting alternate phenylalanine metabolism modules are activated in a Roseburia species-dependent manner.
Variability in cytokine production after BCG vaccination is associated with the abundance of microbial genomes, which in turn affect or produce metabolites in circulation. Roseburia is found to alter both trained immune responses and phenylalanine metabolism, revealing microbes and microbial products that may alter BCG-induced immunity. Together, our findings contribute to the understanding of specific and trained immune responses after BCG vaccination.
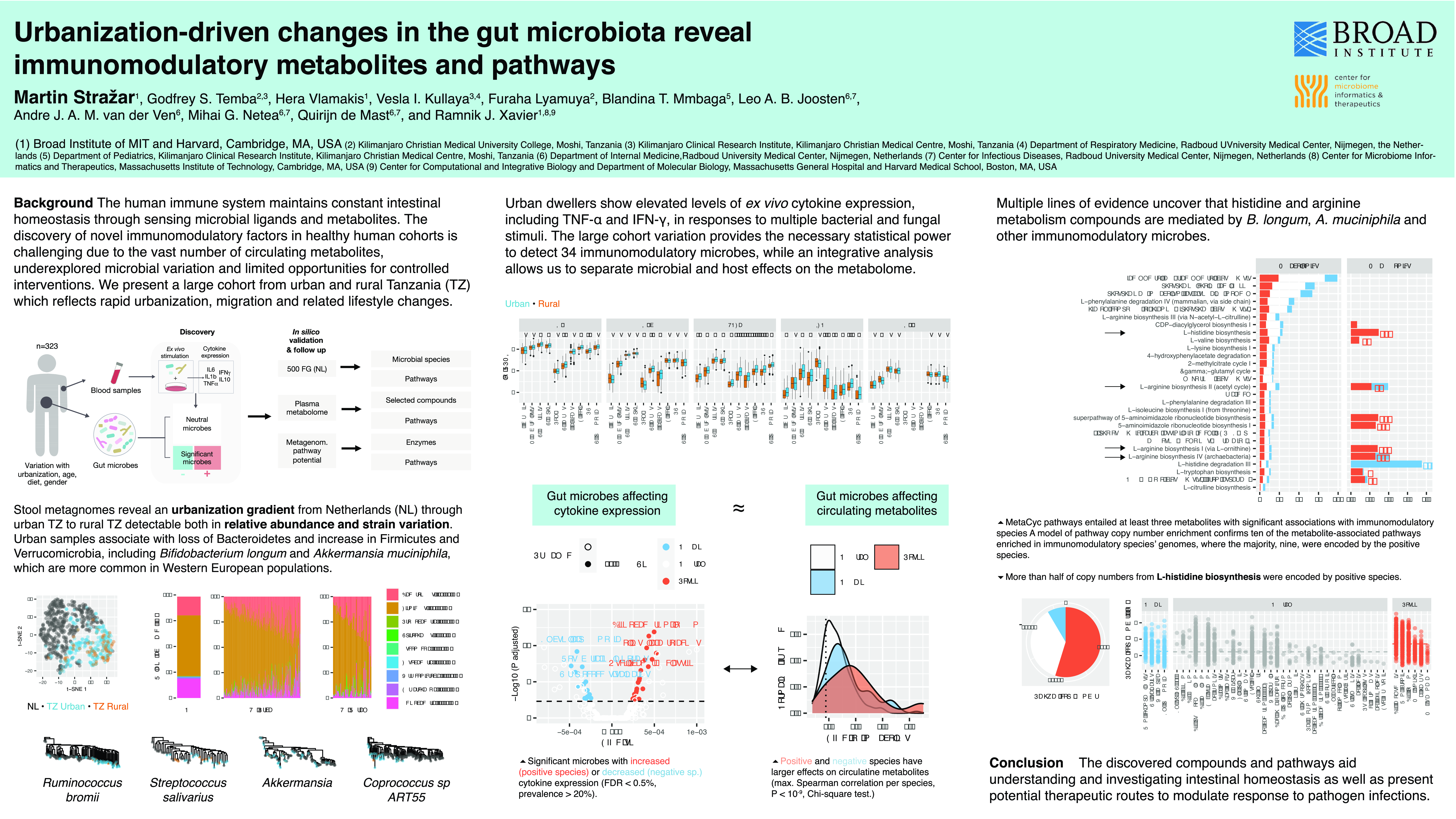
Stražar, M., Temba, G. S., Vlamakis, H., Kullaya, V. I., Lyamuya, F., Mmbaga, B. T., ... Xavier, R. J. (2021). Gut microbiome-mediated metabolism effects on immunity in rural and urban African populations. Nature communications, 12(1), 1-15. Nature Communication Editors’ Highlights pages (showcase of the 50 best papers recently published in an area) | Link | Toggle abstract
The human gut microbiota is increasingly recognized as an important factor in modulating innate and adaptive immunity through release of ligands and metabolites that translocate into circulation. Urbanizing African populations harbor large intestinal diversity due to a range of lifestyles, providing the necessary variation to gauge immunomodulatory factors. Here, we uncover a gradient of intestinal microbial compositions from rural through urban Tanzanian, towards European samples, manifested both in relative abundance and genomic variation observed in stool metagenomics. The rural population shows increased Bacteroidetes, led by Prevotella copri, but also presence of fungi. Measured ex vivo cytokine responses were significantly associated with 34 immunomodulatory microbes, which have a larger impact on circulating metabolites than non-significant microbes. Pathway effects on cytokines, notably TNF-α and IFN-γ, differential metabolome analysis and enzyme copy number enrichment converge on histidine and arginine metabolism as potential immunomodulatory pathways mediated by Bifidobacterium longum and Akkermansia muciniphila.
Poličar, P. G., Stražar, M., and Zupan, B. (2021). Embedding to reference t-SNE space addresses batch effects in single-cell classification. Machine Learning, 1-20. | Link | Toggle abstract
Dimensionality reduction techniques, such as t-SNE, can construct informative visualizations of high-dimensional data. When jointly visualising multiple data sets, a straightforward application of these methods often fails; instead of revealing underlying classes, the resulting visualizations expose dataset-specific clusters. To circumvent these batch effects, we propose an embedding procedure that uses a t-SNE visualization constructed on a reference data set as a scaffold for embedding new data points. Each data instance from a new, unseen, secondary data is embedded independently and does not change the reference embedding. This prevents any interactions between instances in the secondary data and implicitly mitigates batch effects. We demonstrate the utility of this approach by analyzing six recently published single-cell gene expression data sets with up to tens of thousands of cells and thousands of genes. The batch effects in our studies are particularly strong as the data comes from different institutions using different experimental protocols. The visualizations constructed by our proposed approach are clear of batch effects, and the cells from secondary data sets correctly co-cluster with cells of the same type from the primary data. We also show the predictive power of our simple, visual classification approach in t-SNE space matches the accuracy of specialized machine learning techniques that consider the entire compendium of features that profile single cells.
Poličar, P. G., Stražar, M., Zupan, B. (2019). openTSNE: a modular Python library for t-SNE dimensionality reduction and embedding. BioRxiv, 731877. | Link | Toggle abstract
Point-based visualisations of large, multi-dimensional data from molecular biology can reveal meaningful clusters. One of the most popular techniques to construct such visualisations is t-distributed stochastic neighbor embedding (t-SNE), for which a number of extensions have recently been proposed to address issues of scalability and the quality of the resulting visualisations. We introduce openTSNE, a modular Python library that implements the core t-SNE algorithm and its extensions. The library is orders of magnitude faster than existing popular implementations, including those from scikit-learn. Unique to openTSNE is also the mapping of new data to existing embeddings, which can surprisingly assist in solving batch effects.
openTSNE is available at https://github.com/pavlin-policar/openTSNE.
Godec, P., Pančur, M., Ilenič, N., Čopar, A., Stražar, M., Erjavec, A., ... Zupan, B. (2019). Democratized image analytics by visual programming through integration of deep models and small-scale machine learning. Nature communications, 10(1), 1-7. | Link | Toggle abstract
Analysis of biomedical images requires computational expertize that are uncommon among biomedical scientists. Deep learning approaches for image analysis provide an opportunity to develop user-friendly tools for exploratory data analysis. Here, we use the visual programming toolbox Orange (http://orange.biolab.si) to simplify image analysis by integrating deep-learning embedding, machine learning procedures, and data visualization. Orange supports the construction of data analysis workflows by assembling components for data preprocessing, visualization, and modeling. We equipped Orange with components that use pre-trained deep convolutional networks to profile images with vectors of features. These vectors are used in image clustering and classification in a framework that enables mining of image sets for both novel and experienced users. We demonstrate the utility of the tool in image analysis of progenitor cells in mouse bone healing, identification of developmental competence in mouse oocytes, subcellular protein localization in yeast, and developmental morphology of social amoebae.
Stražar, M., Žagar, L., Kokošar, J., Tanko, V., Erjavec, A., Poličar, P. G., ... Zupan, B. (2019). scOrange—a tool for hands-on training of concepts from single-cell data analytics. Bioinformatics, 35(14), i4-i12. | Link | Toggle abstract
Single-cell RNA sequencing allows us to simultaneously profile the transcriptomes of thousands of cells and to indulge in exploring cell diversity, development and discovery of new molecular mechanisms. Analysis of scRNA data involves a combination of non-trivial steps from statistics, data visualization, bioinformatics and machine learning. Training molecular biologists in single-cell data analysis and empowering them to review and analyze their data can be challenging, both because of the complexity of the methods and the steep learning curve.
We propose a workshop-style training in single-cell data analytics that relies on an explorative data analysis toolbox and a hands-on teaching style. The training relies on scOrange, a newly developed extension of a data mining framework that features workflow design through visual programming and interactive visualizations. Workshops with scOrange can proceed much faster than similar training methods that rely on computer programming and analysis through scripting in R or Python, allowing the trainer to cover more ground in the same time-frame. We here review the design principles of the scOrange toolbox that support such workshops and propose a syllabus for the course. We also provide examples of data analysis workflows that instructors can use during the training.
scOrange is an open-source software. The software, documentation and an emerging set of educational videos are available at http://singlecell.biolab.si.
Stražar, M., Curk, T. (2019). Approximate multiple kernel learning with least-angle regression. Neurocomputing, 340, 245-258. | Link | Toggle abstract | Preview
Kernel methods provide a principled way for general data representations. Multiple kernel learning and kernel approximation are often treated as separate tasks, with considerable savings in time and memory expected if the two are performed simultaneously.
Our proposed Mklaren algorithm selectively approximates multiple kernel matrices in regression. It uses Incomplete Cholesky Decomposition and Least-angle regression (LAR) to select basis functions, achieving linear complexity both in the number of data points and kernels. Since it approximates kernel matrices rather than functions, it allows to combine an arbitrary set of kernels. Compared to single kernel-based approximations, it selectively approximates different kernels in different regions of the input spaces.
The LAR criterion provides a robust selection of inducing points in noisy settings, and an accurate modelling of regression functions in continuous and discrete input spaces. Among general kernel matrix decompositions, Mklaren achieves minimal approximation rank required for performance comparable to using the exact kernel matrix, at a cost lower than 1% of required operations. Finally, we demonstrate the scalability and interpretability in settings with millions of data points and thousands of kernels.
Stražar M., Ule J., Žitnik M, Zupan B., Curk T. (2016), Orthogonal matrix factorization enables integrative analysis of multiple RNA binding proteins. Bioinformatics. | Link | Toggle abstract | Preview
RNA binding proteins (RBPs) play important roles in post-transcriptional control of gene expression, including splicing, transport, polyadenylation and RNA stability. To model protein–RNA interactions by considering all available sources of information, it is necessary to integrate the rapidly growing RBP experimental data with the latest genome annotation, gene function, RNA sequence and structure. Such integration is possible by matrix factorization, where current approaches have an undesired tendency to identify only a small number of the strongest patterns with overlapping features. Because protein–RNA interactions are orchestrated by multiple factors, methods that identify discriminative patterns of varying strengths are needed.
We have developed an integrative orthogonality-regularized nonnegative matrix factorization (iONMF) to integrate multiple data sources and discover non-overlapping, class-specific RNA binding patterns of varying strengths. The orthogonality constraint halves the effective size of the factor model and outperforms other NMF models in predicting RBP interaction sites on RNA. We have integrated the largest data compendium to date, which includes 31 CLIP experiments on 19 RBPs involved in splicing (such as hnRNPs, U2AF2, ELAVL1, TDP-43 and FUS) and processing of 3’UTR (Ago, IGF2BP). We show that the integration of multiple data sources improves the predictive accuracy of retrieval of RNA binding sites. In our study the key predictive factors of protein–RNA interactions were the position of RNA structure and sequence motifs, RBP co-binding and gene region type. We report on a number of protein-specific patterns, many of which are consistent with experimentally determined properties of RBPs.
Kavšček, M., Stražar, M., Curk, T., Natter, K., Petrovič, U. (2015). Yeast as a cell factory: current state and perspectives. Microbial cell factories, 14(1), 1 | Link | Toggle abstract | Preview
The yeast Saccharomyces cerevisiae is one of the oldest and most frequently used microorganisms in biotechnology with successful applications in the production of both bulk and fine chemicals. Yet, yeast researchers are faced with the challenge to further its transition from the old workhorse to a modern cell factory, fulfilling the requirements for next generation bioprocesses. Many of the principles and tools that are applied for this development originate from the field of synthetic biology and the engineered strains will indeed be synthetic organisms. We provide an overview of the most important aspects of this transition and highlight achievements in recent years as well as trends in which yeast currently lags behind. These aspects include: the enhancement of the substrate spectrum of yeast, with the focus on the efficient utilization of renewable feedstocks, the enhancement of the product spectrum through generation of independent circuits for the maintenance of redox balances and biosynthesis of common carbon building blocks, the requirement for accurate pathway control with improved genome editing and through orthogonal promoters, and improvement of the tolerance of yeast for specific stress conditions. The causative genetic elements for the required traits of the future yeast cell factories will be assembled into genetic modules for fast transfer between strains. These developments will benefit from progress in bio-computational methods, which allow for the integration of different kinds of data sets and algorithms, and from rapid advancement in genome editing, which will enable multiplexed targeted integration of whole heterologous pathways. The overall goal will be to provide a collection of modules and circuits that work independently and can be combined at will, depending on the individual conditions, and will result in an optimal synthetic host for a given production process.
Stražar, M., Mraz, M., Zimic, N., Moškon, M. (2014). An adaptive genetic algorithm for parameter estimation of biological oscillator models to achieve target quantitative system response. Natural Computing, 13(1), 119-127 | Link | Toggle abstract | Preview
Mathematical modeling has become an integral part of synthesizing gene regulatory networks. One of the common problems is the determination of parameters, which are a part of the model description. In the present work, we propose a customized genetic algorithm as a method to determine the parameters such that the underlying oscillatory system exhibits the target behavior. We propose a problem specific, adaptive fitness function evaluation and a method to quantify the effect of a single parameter on the system response. The properties of the algorithm are highlighted and confirmed on two test cases of synthetic biological oscillators.
Lebar, T., Bezeljak, U., Golob, A., Jerala, M., Kadunc, L., Pirš, B., Stražar M., ... Jerala, R. (2014). A bistable genetic switch based on designable DNA-binding domains. Nature communications, 5 | Link | Toggle abstract | Preview
Bistable switches are fundamental regulatory elements of complex systems, ranging from electronics to living cells. Designed genetic toggle switches have been constructed from pairs of natural transcriptional repressors wired to inhibit one another. The complexity of the engineered regulatory circuits can be increased using orthogonal transcriptional regulators based on designed DNA-binding domains. However, a mutual repressor-based toggle switch comprising DNA-binding domains of transcription-activator-like effectors (TALEs) did not support bistability in mammalian cells. Here, the challenge of engineering a bistable switch based on monomeric DNA-binding domains is solved via the introduction of a positive feedback loop composed of activators based on the same TALE domains as their opposing repressors and competition for the same DNA operator site. This design introduces nonlinearity and results in epigenetic bistability. This principle could be used to employ other monomeric DNA-binding domains such as CRISPR for applications ranging from reprogramming cells to building digital biological memory.
Moškon, M., Zimic, N., Stražar, M., Mraz, M. (2013). Comparison of selected performances of biological and electronic information processing structures. Przegląd Elektrotechniczny, 89 | Link | Toggle abstract | Preview
We present the information processing perspective on biological systems. Several metrics, similar to the ones used in digital electronic circuits, are introduced. These metrics allow us to compare biological information processing structures with their electronic counterparts, to define the ones with the best dynamical properties, analyse their compatibility and most importantly, automatize their design. Regarding the metric values obtained and used on a simple example, target applications of synthetic information processing biological structures are discussed.
Conference presentations
Presentations at conferences and invited talks
Stražar, Martin. Gut microbial metabolites as effectors and markers in chronic inflammatory disease. V: Bageco 2025 : 17th Symposium on Bacterial Genetics and Ecology : 1-4 July 2025, Graz, Austria.
Stražar, Martin. Antigen discovery and spatial microbiome sequencing tools dive deep in the mucosa : Invited lecture at 10th Theodor Escherich Symposium, Med Uni Graz, 18. & 19. January 2024.
Stražar, Martin (2023). HLA-II immunopeptidome profiling and deep learning reveal features of antigenicity to inform antigen discovery. Massachusetts General Hospital, MolBio Seminar, April 7, 2023.
Stražar, Martin (2022). Gut microbiome-mediated metabolism effects on immunity in rural and urban African populations. Ithaca: Cornell University, September 30, 2022.
Godec, P., Zupan B., Tanko V., Stražar M. (2022), Efficient Matching of Single Cells and Cell Types, XAI-Healthcare eXplainable AI in Healthcare, 1-day workshop June 11-14, 2022 in conjunction with IEEE ICHI 2022, Rochester, Minnesota, USA
Stražar, M., Temba, G. S., Vlamakis, H., Kullaya, V. I., Lyamuya, F., Mmbaga, B. T., ... Xavier, R. J. (2021). Urbanization-driven changes in the gut microbiota reveal immunomodulatory metabolites and pathways,
- Impact of Environmental Exposures on the Microbiome and Human Health, NIEHS Workshop, February 23, 2021. Virtual.
- Harnessing the Microbiome for Disease Prevention and Therapy, Keystone Symposium, January 18-20, 2021. Keystone, Co. USA.
{kind=link}
Stražar, M. and Žagar, L. and Kokošar, J. and Tanko, V. and Poličar, P. and Erjavec, A. and Starič, A. and Menon, V. and Chen, R. and Shaulsky, G. and Lemire, A. and Parikh, A. and Zupan, B. (2018). scOrange: Single-Cell Data Mining for Everyone. The 16th International NETTAB workshop. October 22-24. Genova, Italy. | PDF | PNG
{kind=link}
Stražar M., Ule J., Curk T. (2017). csDEX: condition-specific differential exon expression. 25th annual International Conference on Intelligent Systems for Molecuar Biology. European conference on Computational Biology. Prague, Czech Republic | PDF | PNG
{kind=link}
Stražar M., Curk T. (2016) Learning the kernel matrix by predictive low-rank approximations. Machine Learning Summer School. Cadiz, Spain | PDF | PNG
{kind=link}
Stražar M., Žitnik M., Zupan B., Ule J., Curk T. (2015). Orthogonal nonnegative factorization-based analysis of nineteen protein-RNA interaction CLIP data sets. 23rd annual International Conference on Intelligent Systems for Molecuar Biology. European conference on Computational Biology. Dublin, Ireland | PDF | PNG
{kind=link}