Molecule predictions with chemical formula transformers
Metabolism, the process to extract energy from molecules, is central to all life. The ability to break down and synthesize molecules enables life forms to continuously regenerate and adapt to environments characterized by availability of chemical species. Metabolomics methods study molecular composition of various biological samples, from human tissues to bacterial cultures. Knowing which molecules make up these systems is crucial to understand how they function in normal or disease states.
Capturing molecular compositions happens at one of the smallest scales and, roughly speaking, works by separating and weighing different masses by processes of chromatography and mass spectrometry. Although the profiles of masses and their abundances are measured very precisely, they most often lack sufficient information to truly identify molecules. As atoms can form molecules of many different arrangements, a single mass can pertain to many different molecules from the same atoms.
While we cannot see the molecules, we can experimentally force bond breaking, causing them to form fragments. To identiy molecules with greater precision, we exploit the fact that some molecular fragments are more likely to occur then others. This is the reasingin behind tandem mass spectrometry (MS/MS), where masses are isolated and exposed to inert gases, where collisions force bonds inthe molecules to break. The result is a list of observed fragment masses and their intensities, a large boost in available information allowing us to unmask the culprit molecule.
While often still done manually, the task largely became the domain of computational prediction methods, where machine learning is to a large extent taking over the decision process. Our latest metabolite prediction framework, Metabolite Inference with Spectrum Transformers (MIST) was developed by Sam Goldman, supervised by Connor Coley (MIT Chemical Engineering) and is available in open source (see below).
It is the latest in the field of MS/MS prediction tools and combines modern practical advances in deep learning, including graph neural network, attention mechanisms, and clever training which includes data augmentation. While most traditional machine learning methods work with numerical vectors of fixed dimension, MIST represents input data points as graphs of molecular formulae. Since different molecules break down into different numbers of fragment, this “bag-of-formula” representation provides a description more representative of real world entities. This makes sense, since fragments break down into further smaller fragments in a recursive manner.
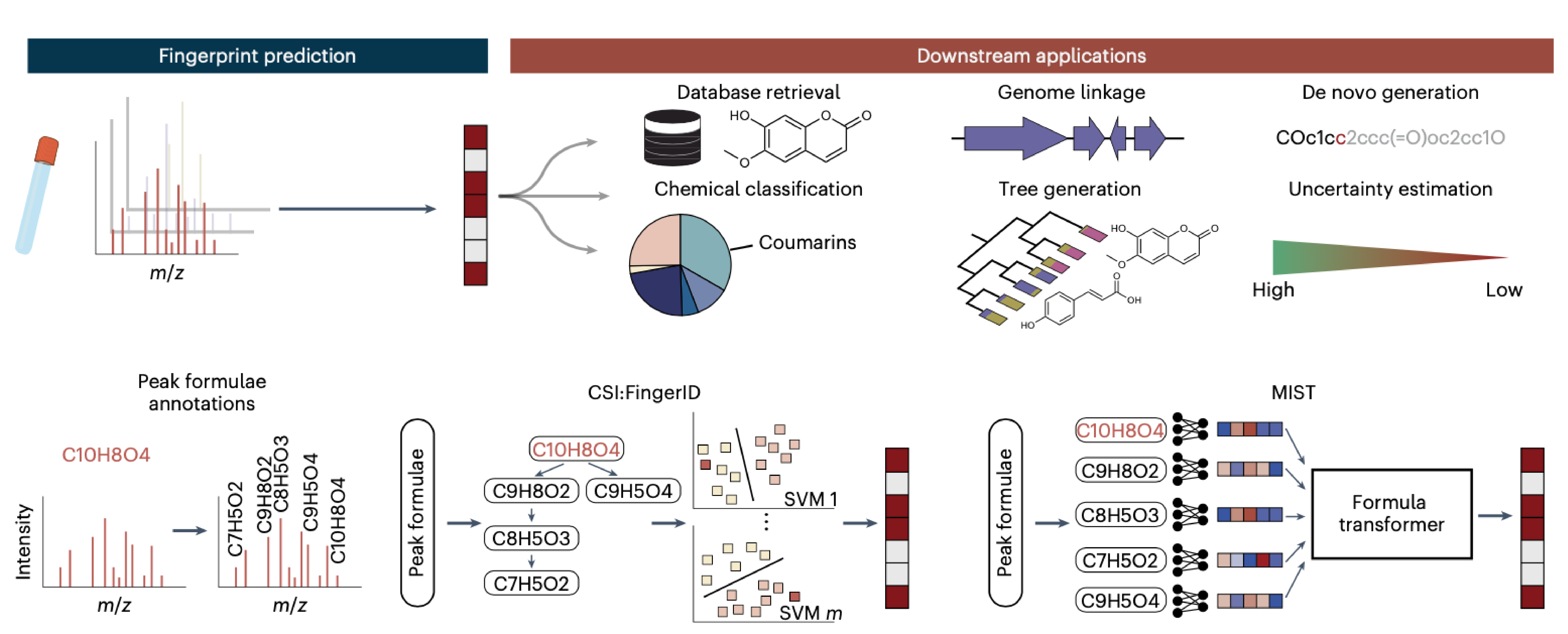
The methods outperforms established MS/MS predictions methods based on traditional machine learning for multiple molecular classes. In practice, it has been used to identify many novel molecules characteristic for chronic inflammation of the intestines, where presence of harmful bacteria cause unexpected breakdown of host proteins. Here, MIST was used to identify novel candidate dipeptide-like molecules, which are about to be investigated further and may themselves be of bacterial origin.
While molecule prediction from mass spectra is becoming an established toolbox of many investigations, we are merely scratching the surface of treasures to be found in bacteria, including novel metabolites, natural products with antimicrobial activity or drug-like molecules.
Learn more in Nature Machine Intelligence or try out MIST on GitHub.